
.png)
This blog is meant to be a resource for physicians treating patients with Interstitial Pneumonia during the COVID-19 pandemic. This information has been pulled from our 18th Edition Internal Medicine Core.
Overview of Idiopathic Interstitial Pneumonias
We will now discuss the second category of ILDs— idiopathic interstitial pneumonias (IIPs). Here is a list of IIPs in the order of occurrence (high to low):
- Idiopathic pulmonary fibrosis (IPF; usual interstitial pneumonitis [UIP] is the prototype)
- Nonspecific interstitial pneumonia (NSIP)
- Cryptogenic organizing pneumonia (COP, an idiopathic form of organizing pneumonia)
- Acute interstitial pneumonia (AIP)
- Respiratory bronchiolitis-associated ILD (RB-ILD)
- Desquamative interstitial pneumonia (DIP)
- Lymphocytic interstitial pneumonia (LIP)
Each of these entities has specific histopathologic findings. IPF also has typical clinical and radiologic findings.
Cryptogenic Organizing Pneumonia
Cryptogenic organizing pneumonia (COP) is an idiopathic form of organizing pneumonia. COP is a bronchiolitis (inflammation of the small airways) and a chronic alveolitis (the organizing pneumonia). The bronchiolitis causes a proliferation of granulation tissue within the small airways and alveolar ducts.
Consider COP in a patient with an insidious onset (weeks to 1–2 months) of cough, fever, dyspnea, malaise, and myalgias. Often, patients have had multiple courses of antibiotics without effect. Rales are common. Chest x-ray shows some interstitial disease, bronchial thickening, and patchy bilateral alveolar infiltrates. Chest CT shows bilateral, peripheral, and often triangular infiltrates, but these can become confluent. PFTs demonstrate a restrictive pattern with a reduced diffusion capacity.
You must differentiate COP from IPF because, contrary to IPF, COP has a good prognosis and a good response to steroids. To differentiate IPF from COP, know that IPF is even more insidious in onset (> 6 months) and the patients do not have fever (Table 6-9 on page 6-32). Radiographically, the consolidative infiltrates of COP differ from the subpleural reticulations and fibrosis with traction bronchiectasis and honeycombing of UIP. Lung biopsy is the definitive means of diagnosing COP.
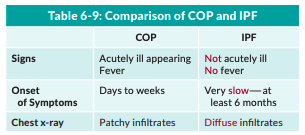
Table 6-9: Comparison of COP and IPF
Corticosteroids are the treatment of choice for COP. COP does not respond to antibiotics. Slowly taper corticosteroids over 6–12 months because exacerbations can occur with rapid tapering. Corticosteroid-sparing treatment can be used—typically cyclophosphamide for refractory cases requiring high doses of steroids.
Check out the next blog in this series: IM Core Excerpt: Acute Respiratory Distress Syndrome
